- Client S/W for installation
- Upload FASTQ + Analyze automatically
- Download QC report, VCF files
- Web-based platform for mass sample analysis
- You can make desired profiles
- Various options are available
- Download VCF, gVCF, bam, QC report, annotation
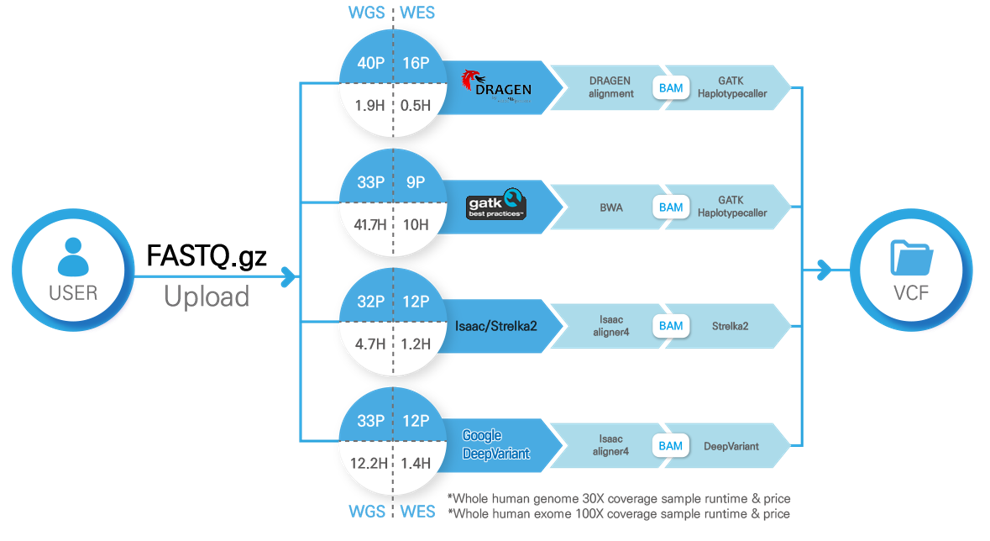
Price and Runtime by 30X coverage
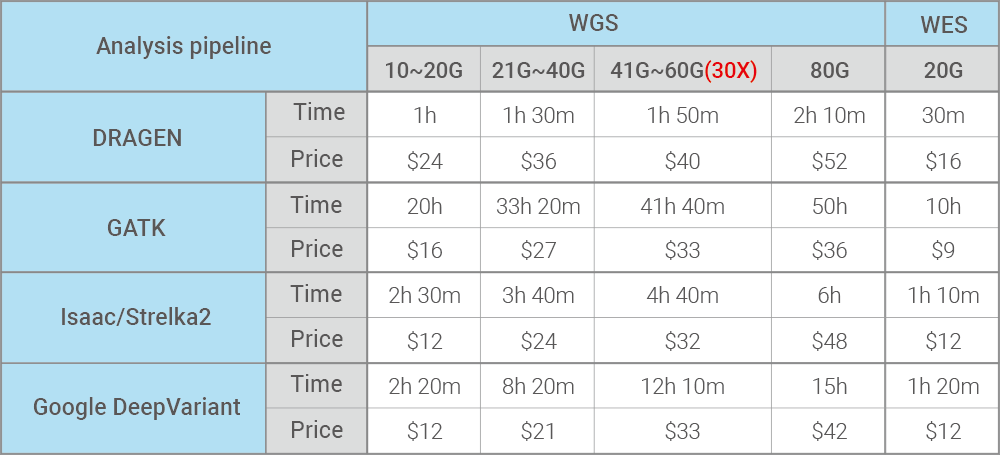
Price and Runtime by FASTQ file size
-
1. GATK4 Best Practices
This analysis tool uses the open-sourced Genome Analysis Toolkit 4 from Broad institute.
The pipeline in this tool is based on the GATK Best Practices Workflow for performing variant discovery analysis in NGS data.
Mapping to a reference genome is performed by BWA and the calling SNPs and indels is achieved by using the GATK HaplotypeCaller. -
2. DRAGEN analysis
Illumina DRAGEN Complete Suite is an application that provides a comprehensive pipeline package for analyzing NGS data.
The DRAGEN GATK Best Practices pipeline, used in this app, takes advantage of DRAGEN's ultra-fast analysis method to speed up the open-sourced GATK 4.0 Haplotype Variant Caller.
This pipeline contains all required analysis steps (mapping / aligning, position sorting, duplicate marking, and variant calling) specified by the Broad Institute.
The advantages of DRAGEN are ultra-rapid speed, cost savings and high accuracy. -
3. Isaac/Strelka2 analysis
This analysis mode uses Isaac aligner4 for alignment and uses Strelka2 for variant calling.
Isaac aligner4 is a new high-speed read mapping tool included in the illumina hiseq analysis software. It provides read mapping, sorting, PCR duplicate removal, and bam file generation into one optimization task.
The generated bam file is used to generate a VCF file by the Strelka2 Germline variant caller.
Strelka2 is a fast and accurate small variant caller optimized for analysis of germline variation in small cohorts and somatic variation in tumor/normal sample pairs. -
4. DeepVariant analysis
This analysis mode uses Isaac aligner4 for alignment and uses Google DeepVariant for variant calling.
Isaac aligner4 is a new high-speed read mapping tool included in the illumina hiseq analysis software. It provides read mapping, sorting, PCR duplicate removal, and bam file generation into one optimization task.
DeepVariant is an analysis pipeline that uses a deep neural network to call genetic variants from aligned reads.
For technical details describing how DeepVariant works please see the paper Nature Biotechnology publication.